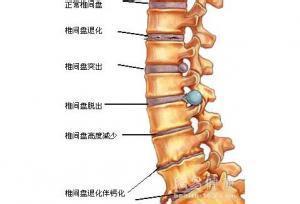
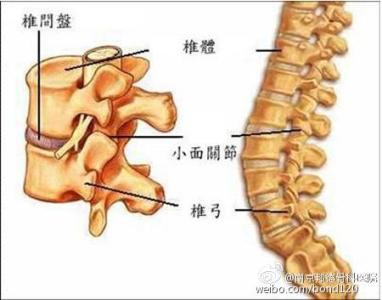
无论是发病机制还是治疗手段,肌萎缩侧索硬化(ALS)都是一种难以捉摸的疾病。这种疾病的异质性对于人们开发有效疗法造成了阻碍,但也因此道出了根据每名患者的情况进行个体化治疗的需求。
遗传学和细胞生物学方面的研究进展已经阐明了一些公认的关键致病机制,对于未来的疾病修饰治疗开启了一扇美好的窗户。关于ALS这一疾病遗传原因方面的知识景观正在迅速丰富,并且在2015年,一些新的基因,如CHCHD10、NEK1和TBK1纷纷在舞台上登场。
其中,TBK1看上去尤其有趣,既考虑了用于识别ALS的GWAS(全基因组关联研究)新两步法,又提示了可能的机制。Cirulli等人[1]对2869名白人ALS患者和6405名对照组进行了全外显子组测序。TBK1与这一疾病相关,加之几个基因已被证实与ALS存在关联(分别为SOD1、VCP、TARDBP),确定了这一研究的质量。TBK1编码TANK结合激酶1,是一种参与自噬和炎症的蛋白质,已提出两种关于ALS的致病机制。有趣的是,TANK结合激酶1与视神经病变诱导基因和P62结合并磷酸化,两种蛋白质分别由ALS相关基因OPTN和SQSTM1编码,并且似乎是沉积体途径的一个重要组成部分,为清除病理性核蛋白夹杂物所必需。TBK1基因突变的患者存在不同表型,包括单纯运动ALS、额颞叶痴呆(FTD),以及ALS-FTD。
解开ALS潜在的分子机制另一项重要的步骤,是在两个独立的核转运缺陷组中,鉴定了C9ORF72基因第一内含子中携带有GGGGCC(G4C2)六核苷酸重复扩张。这种遗传缺陷在50%的家族性ALS和5%的散发型ALS中可见。Zhang等人[2]及Freibaun等人[3]鉴定了RanGAP,一种在果蝇模型中与G4C2相互作用的蛋白质,作为核浆转运的调节关键,作用于核孔的外部水平。RanGAP的核转运在C9ORF72重复扩张的果蝇模型中被修改,并已成功通过反义寡核苷酸治疗获救。这种新型致病机制为ALS的治疗提供了一个充满希望的新途径。
神经肌肉退行性疾病的致病机制和推定治疗途径鉴定方面的进一步进展预计将来自RNA转录调控的分析。RNA加工机制的改变引起多种疾病的发生,包括神经变性和线粒体疾病。由TARDBP基因突变造成的ALS和脊髓肌肉萎缩1型和2型是与RNA剪切改变有关的疾病例子。RNA剪切的改变在允许单个基因的多种蛋白质进行编码方面扮演了重要的组成部分。蛋白质组不断扩大的这种机制具有巨大的生物影响和高水平的复杂性。
Xiong等人[4]使用原始机器学习生物信息学的方法,发现超过2万个单核苷酸变异(SNV)位点,包括错义、无义、甚至是同义SNV,可以调节细胞-特异mRNA的外显子数目。作者成功地证明了他们的新型脊髓肌肉萎缩计算模型,是童年第二个最常见的常染色体隐性遗传病和婴儿期死亡的主要原因之一。这一可靠的预测模型通过对新的致病性RNA剪切改变的鉴定,扩展了全基因组关联研究的潜力。

蛋白质合成和降解之间平衡的改变导致错误折叠的蛋白聚集体在细胞内积累及清除机制失效,这是在ALS和腓骨肌萎缩神经病变中出现的机制。内质网应激在通过折叠蛋白反应的活化调节蛋白质稳态方面具有关键作用。针对错误折叠蛋白积累的一线适应性反应为真核翻译起始因子2(eIF2a)的磷酸化,导致蛋白质合成减少。这种自我限制响应的增强对于抢救错误折叠蛋白积累的细胞十分重要。
Das等人[5]的研究证实,sephin1选择性结合并抑制eIF2a磷酸酶的调节亚基,在超氧化物歧化酶1相关ALS和髓鞘蛋白0相关腓骨肌萎缩(CMT1B)的转基因小鼠模型中,安全地防止了分子、形态和运动功能障碍。这些发现同时提供了ALS和CMT1B疾病修饰治疗的特定基础,为与错误折叠蛋白的积累有关的多种疾病治疗开启了新的道路。
延伸阅读:牛奶可引起帕金森样神经变性 或因农药污染出生前的突变可能妨碍心脏和神经系统发育老年神经用药市场年增28%新研究发现多个大脑神经细胞新类型小诊所把三叉神经痛当牙痛治 女子3年拔掉满口牙成年失明者重获视力 视神经学带来新光明