25岁小伙突发胸闷,到医院一检查发现自己竟患有先天性的马凡氏综合征,这是一种遗传性疾病,患者因心血管病变而随时有猝死的危险。确诊后,医院立即对他进行了手术,成功救治了这名患者,这是省内首例通过这项技术将患者救治成功的案例。
25岁的即墨小伙马强(化名)身高1.94米,四肢细长,手指、脚趾似蜘蛛腿,细得让人担心。今年2月份,他突然感觉胸闷、憋气,起初并没有放在心上。
2月底,他突然胸闷加重,便到即墨一家医院就诊,经过检查,发现马强的主动脉有夹层,医生怀疑马强患有先天性马凡氏综合征,无法在当地医院救治,马强随即被转到齐鲁医院青岛院区。
“入院检查后发现这个患者主动脉扩张,普通人的主动脉直径约3厘米,而这个患者的主动脉直径已经达到7厘米,形成主动脉瘤,病情危急,随时有破裂的危险,患者随时可能因大出血或者冠性动脉被压住导致心梗而死亡。”齐鲁医院青岛院区心外科副主任孙文宇告诉记者,。
马凡氏综合征是一种遗传性结缔组织疾病,患者四肢细长,95%的患者存在不同程度的心血管病变,这种病发病急、病情凶险,有50%的病人在发病后72小时内死亡,因此必须在最短的时间内进行手术。
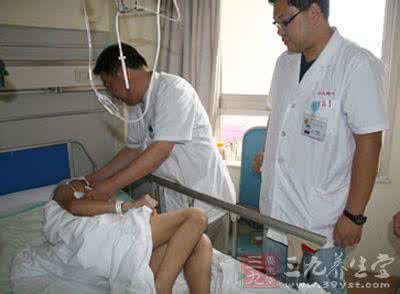
2月28日,历时14个小时,马强的手术最终完成,心脏瓣膜成功置换,这也是省内首例通过血液转流技术将主动脉弓重建、Bentall手术与象鼻支架置入术三台手术合并完成的案例。3月17日,记者在齐鲁医院见到了马强,手术后他恢复顺利,现在病情平稳,已经可以出院了。
马凡氏综合征
马凡氏综合征是一种遗传性结缔组织疾病,为常染色体显性遗传,患病特征为四肢、手指、脚趾细长不匀称,身高明显高于常人,伴有心血管系统异常。发病率为十万分之一到万分之一。
调查显示,有三分之一的马凡氏综合征病人死于32岁以前,三分之二死于50岁左右,平均死亡年龄为40岁,死亡的主要原因是心血管病变,最常见的是主动脉瘤破裂、心包压塞。
延伸阅读:国际罕见病日 盼罕见病进大病医保黄如方称我国罕见病“缺医少药”问题突出研究称基因组编辑为罕见病带来希望罕见病女童服廉价药致全身长黑毛河南辉县2岁罕见病患儿为治病几乎没回过家宁德一男婴患罕见病只吐不拉